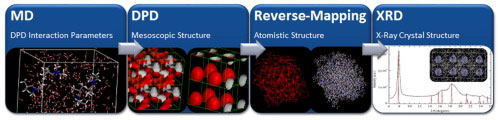
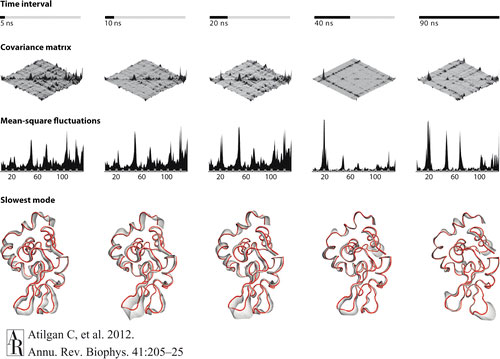
Our goal is to answer the why question instead of the how question for the observed behavior of materials using computational techniques as the tool. Our focus is on the dynamics of complex molecular systems. We address understanding of the working principles of molecular machines. We study organic self-assembled molecules in general, and proteins in particular. We utilize atomistic and coarse-grained simulations supplemented by theoretical developments to cover the pico-millisecond time scales, and nano-micrometer length scales. We have developed network-based models to study the basic features contributing to structure/function relationships in these systems. These include:
Optimal paths along the networks to identify key locations participating in their proper functioning. (available as a server under Services link at //midst.sabanciuniv.edu)
System identification based on perturbation/response scanning (PRS) to study the near-equilibrium dynamics. (PRS server is located at //midst.sabanciuniv.edu/prs)
Prediction of relaxation times contributing to equilibrium fluctuations.